Canonical layout
<SET_NAME>\t<DESCRIPTION_OR_URL>\t<GENE_1>\t<GENE_2>\t...\t<GENE_N>
- SET_NAME: short identifier for the set (no tabs). Prefer A-Z, 0-9,
_, -.
- DESCRIPTION_OR_URL: free text, a reference, or a link. Can be empty (
- or blank after the tab).
- GENE_i: one identifier per column (e.g., HGNC symbols). All sets in a file should use the same ID system and species.
Minimal working example
TNF_SIGNALING\tKEGG hsa04668\tTNF\tTNFRSF1A\tTRADD\tTRAF2\tMAP3K7\tIKBKB\tNFKBIA\tRELA
OXPHOS_CORE\tReactome R-HSA-1428517\tNDUFS1\tNDUFA9\tUQCRC1\tCOX4I1\tATP5F1A
Encoding & line endings
- Use UTF-8 text, Unix line endings (
\n).
- Do not wrap lines; one line = one gene set.
Common pitfalls & how to avoid them
- Mixed identifier namespaces: Only use the official gene symbols (for example from HGNC or MGI).
- Whitespace separators: tabs only between fields. Do not use spaces or commas to separate fields.
- Empty or duplicate genes: drop NA/blank entries and de-duplicate within each set.
- Oversized / undersized sets: extremely large (>5000 genes) or tiny (<3 genes) sets can distort statistics.
- Ambiguous set names: avoid tabs, newlines, or excessive punctuation in
SET_NAME. Keep names unique.
- Inconsistent casing: if you choose HGNC symbols, keep them uppercase (e.g.,
TP53), not mixed case.
Tip: If your source data uses multiple ID types, normalize them first (e.g., map Ensembl → HGNC) before writing the GMT.
Preparing a custom library
- Choose an identifier scheme (e.g., HGNC for human). Good practice is to keep every term in your library using the same scheme.
- Build gene set membership: Create a set of genes for each term.
- Write the GMT with one term per line, tabs between fields, no trailing tabs.
- Validate with the checklist below.
Validation checklist
- All lines have ≥3 columns (name, description, ≥1 gene). Description can be empty but the tab must be present.
- No duplicate
SET_NAME values.
- No duplicate gene entries within a set.
- No tabs inside gene symbols or set names.
- Consistent species and gene symbol namespace.
Quick one-liner to spot non-tab separators (Unix)
grep -n " " your_library.gmt # flags space runs; fix to tabs
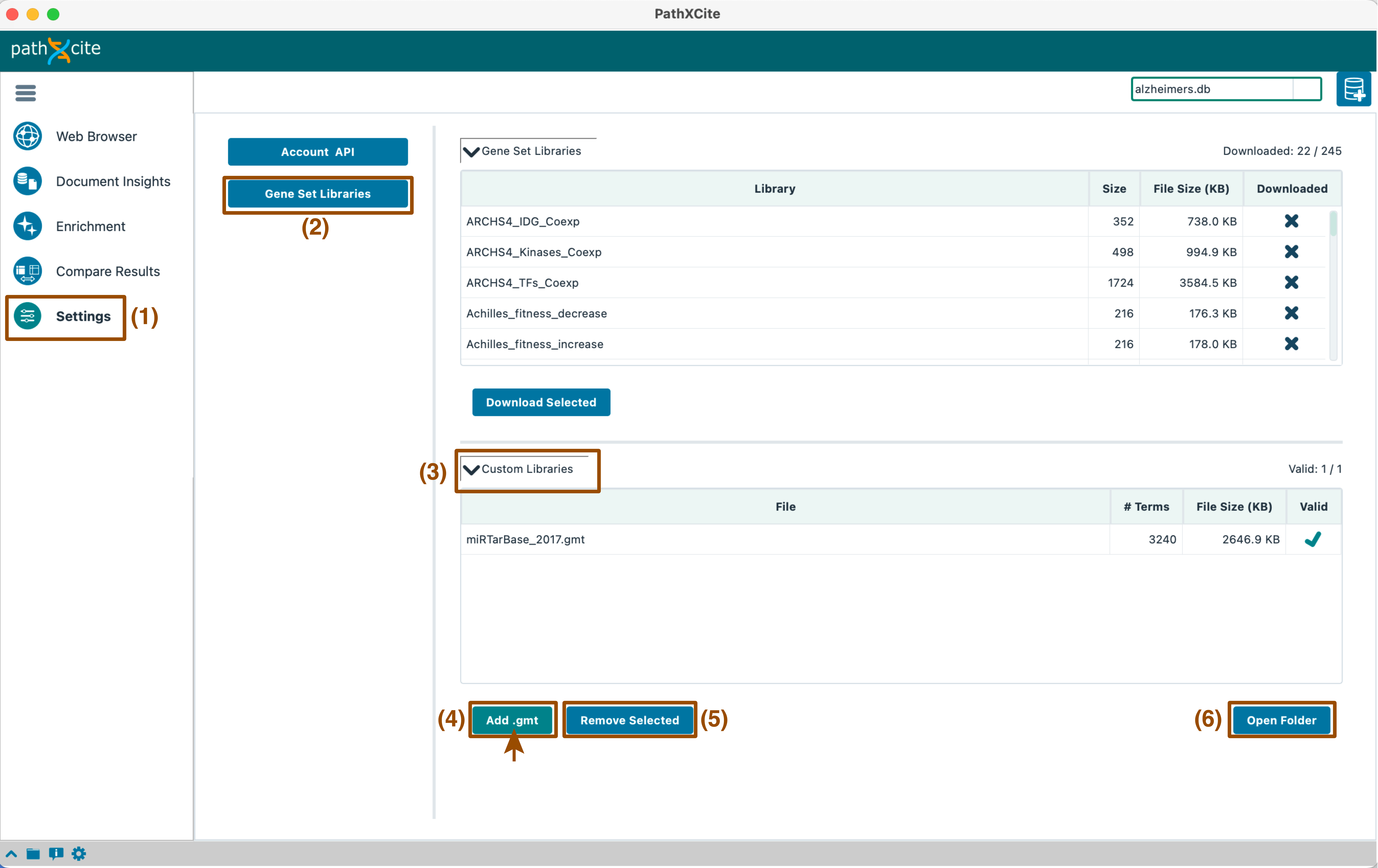
Adding custom libraries to pathXcite
You can add custom libraries either via the UI or by placing files in the data folder.
- Open Settings (1) → Gene Set Libraries (2).
- Scroll to Custom Libraries (3) (bottom panel). Click the arrow to expand if not visible.
- Click Add .gmt and select your
.gmt file.
- Confirm. The table lists name, term count, and file size as well as whether the file has a valid format.
- Click Remove Selected (5) to remove a custom library.
- Click Open Folder (6) to see where the files are stored.
File naming: keep filenames concise (e.g., my_study_markers_hgnc.gmt). Avoid spaces; use - or _.
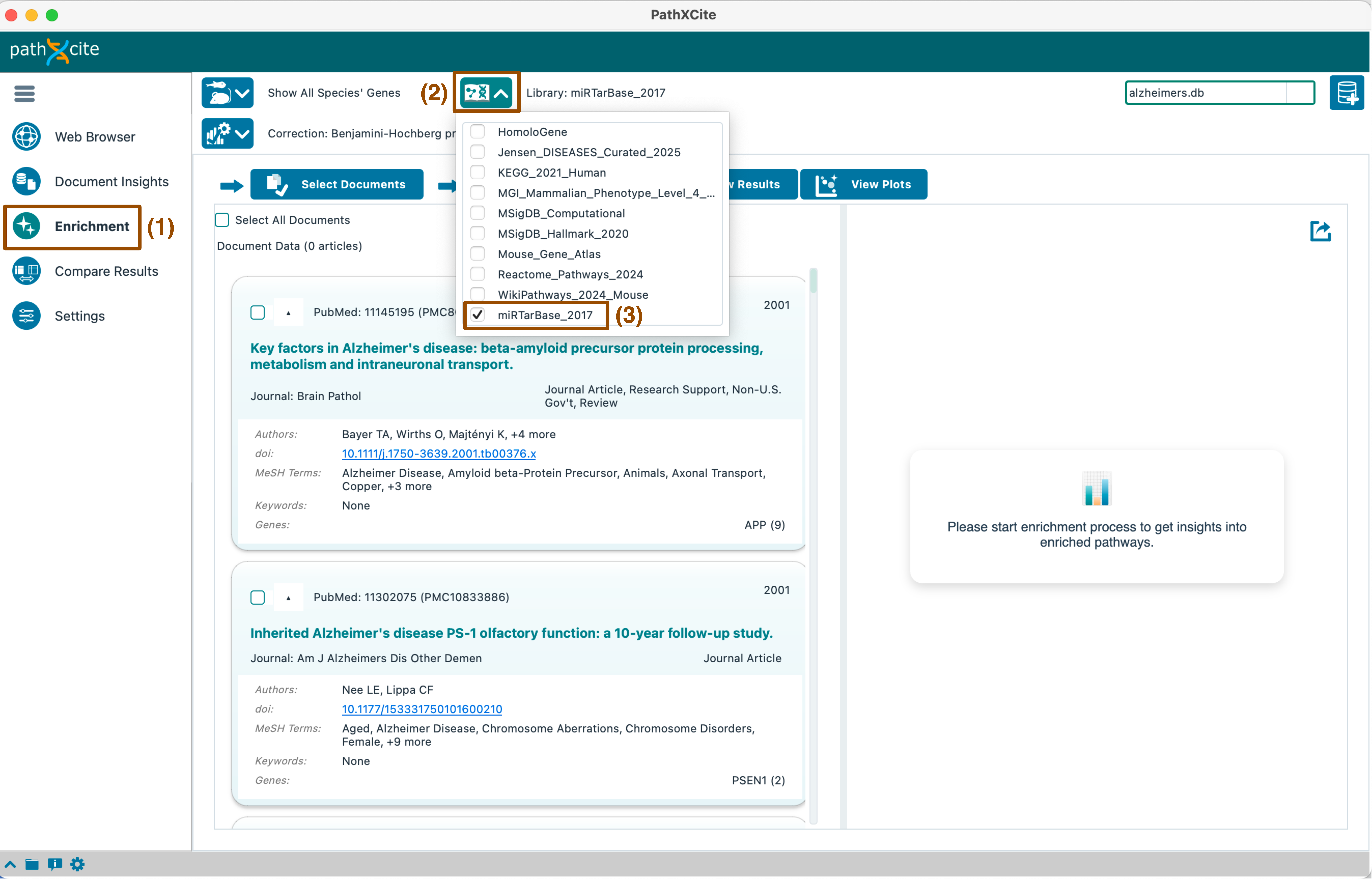
Using your custom library in analyses
- Open the Enrichment (1) module.
- Click on the library selector (2).
- In the library selector, you should now find the custom GMT (3).
Versioning for reproducibility
When a custom GMT underpins key results, archive the exact file with your project and note the download / creation date, species, and ID scheme.
FAQ & troubleshooting
- My library loads but shows zero terms. Check for spaces instead of tabs; ensure each line has at least three fields.
- Some genes look missing. Verify your identifier scheme matches your input list (e.g., HGNC vs Ensembl). Convert if needed.
- Duplicate set names. Ensure unique
SET_NAME per row; rename with a suffix (e.g., _v2).
- Very large sets dominate results. Consider filtering sets to a reasonable size range (e.g., 5-2000 genes) before analysis.
Done! Your custom GMTs are now available.